The website eu-mdr.eu is intended to help companies from different sectors and with different products to reach the safe haven of MDR conformity in an efficient way.
CASE Study 1 - Dental Instruments company - MDD and MDR Compliance Gaps
Type of device: Class 1 instruments - I, Im, Ir ,Irm
Challenge: devices were commercialized without proper documentation. Only under own declaration of conformity. Manufacturer needs to keep device on the market but has not quality system in place and no suitable technical documentation
Support steps:
- Gap analysis to understand current situation and compliance gaps in reference to MDR.
- Definition of action plan per every category of device
- Setting up of missing technical documentation
- Check NB situation. Only 2 notified bodies are currently designated for MDR. Client has not certified them and is not yet in contact with a notified body
- Client has no ISO 13485:2016 certification in place. So different solution /route of conformance were evaluated
- To make sure product could stay on the market it was decided to first properly certify under MDD in order to have certificate valid for 4 years, and in parallel work on update according MDR
CASE Study 2 - Trauma Implant company - Resell under OEM/Virtual Manufacturing
Type of device: Class IIB Implants
Challenge: The company was previously only selling products and wants now to resell under own brand implant from other manufacturer and also develop new ones
Support steps:
- Gap analysis to understand current situation and compliance gaps in reference to MDR.
- Definition of action plan per every category of device
- Support in selecting proper supplier certified with a reliable notified body
- Review of supplier technical file to highlight gaps and estimate efforts
- Creation of technical file for the virtual manufacturer
- Creation of compliant ISO 13485:2016 quality system
- Alignment with notified body
Further information:
To support our clients going trought the MDR path and navigate safe to reach the harbour of compliance we have developed the website eu-mdr.eu.
This website contains a list of free resources and a central repository for MDR related topics and guidance.
Moreover a free Gap Analysis document tool is also available for download.
Here below the KEY POINTS for a Smooth Transition which are also discussed in the Gap Analysis tool:
- Reclassification--> Check immediately if any device will be affected by reclassification
- Select/confirm Notified Body--> Evaluate any notified body change/shift
- UDI and EUDAMED --> Define and plan UDI and EUDAMED activities
- Post-Market Surveillance--> Review compliance of your QMS for PMS activities
- Planning --> If not aleady done, you should start create a MDR Transition Plan immediately to check/review your gaps, split the task into priority list and start planning implementation action
Planning
Manufactures should start an evaluation plan of all their current CE-marked devices in relation to the new MDR guidelines. For some devices, this also means taking into account changes in the classification.
We can support in create a proper Gap Analysis and your custom MDR Transition plan.
The MDR Transition Plan will give you the full picture and overview of your current situation and gaps that needs to be filled to stay compliant.
This will help your company define a proper structured plan, set priority, initiate critical activities on time and smoothly manage the transition.
| Analyze and Asses |
Sub-Task |
Plan and Execute |
Review and Finalize |
| We will start with a gap assessment and evaluation of your current situation considering the type of product and define the required step to achieve compliance to MDR 2017/745. |
The work package will be spitted in different sub task |
A plan will be defined considering priorities and risk and executed |
Each sub task will be reviewed until finalization |
Reclassification
Changes will be made to the way in which medical devices will be classified, with increasingly specific requirements based on the risk to which the patient is exposed. It follows a more restrictive classification and therefore an increase in class for many devices (for example, some devices that come into contact with the spinal cord will pass from class II to class III).
The reclassification of the devices will require costly certification processes for new products and a re-certification of products already on the market. Having said this, it should not be taken for granted that for a device that has not been reclassified the steps to make sure it remains compliant will be quick or simple.
Understanding what has changed between directives 93/42 / CEE medical devices and 90/385 / EEC active implantable medical devices and the new MDR is essential as it is highly probable that a revision of Annex I will be required for most devices, General safety and performance requirements, which will identify new conditions to be applied for each device.
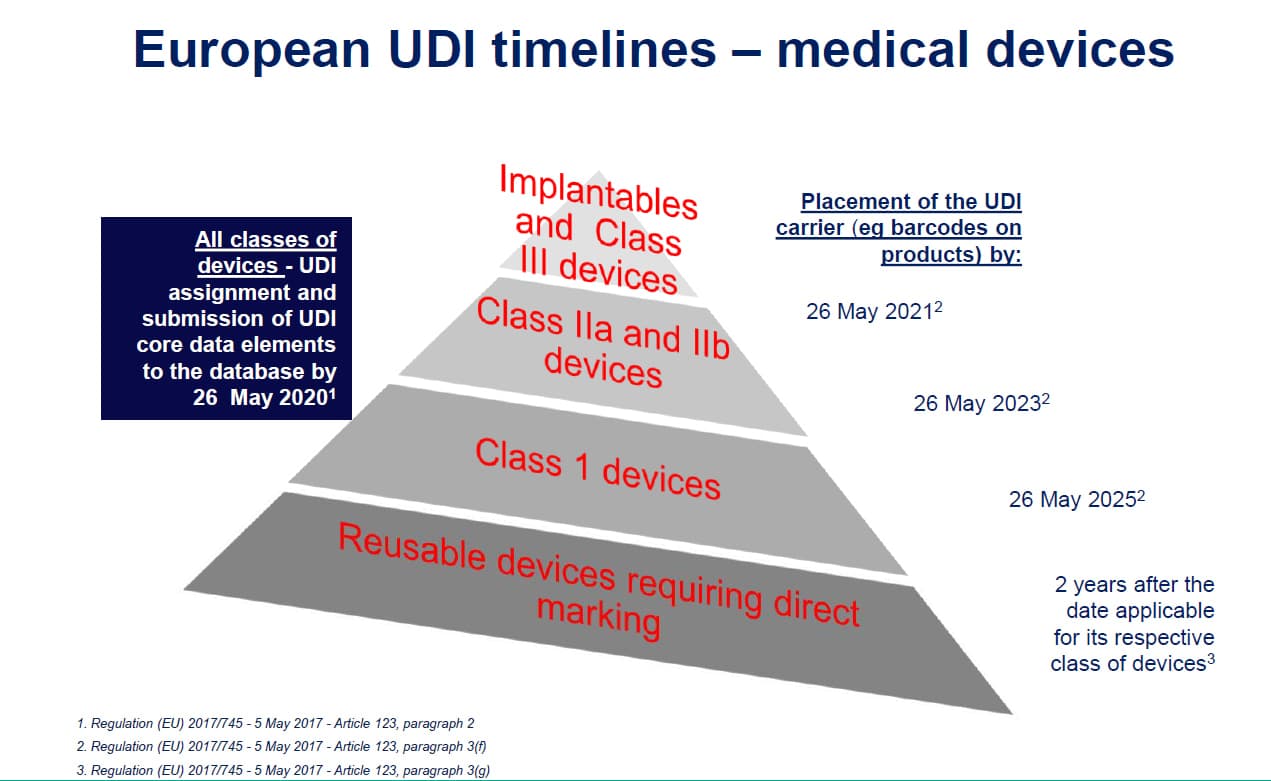
UDI
Apply to all medical devices placed on the market except custom-made devices.
Use:
- Basic UDI-DI is the access key for device-related information entered in EUDAMED
- Reference to Basic UDI-DI in key documentation (Declaration of conformity, certificates)
- UDI shall be used for reporting serious incidents and field safety corrective actions
- UDI storage obligations for Class III implantable devices
UDI issuing entities
The European Commission shall designate by means of implementing acts UDI issuing entities provided that they satisfy certain criteria.
Until the Commission has designated UDI issuing entities, GS1, HIBCC, ICCBBA shall be considered as designated issuing entities.
Assignment/Submission of UDI data/UDI carrier
- Before placing a device on the market, the manufacturer shall assign to the device and – if applicable – to all higher levels of packaging a UDI.
- The UDI carrier shall be placed on the label of the device and on all higher levels of packaging. Higher levels of packaging do not include shipping containers.
- Before a device is placed on the market the manufacturer shall ensure that the information referred to in Part B of Annex V of the device in question is
correctly submitted and transferred to the UDI database
- In some cases, the manufacturer is required to assign a Basic UDI-DI to the device before applying for the conformity assessment (the Notified Body will include a reference to the BASIC UDI-DI in the certificate).
Placement of the UDI carrier:
- Implantable devices and Class III devices (and Class D IVDs): 1 year after the date of application.
- Class IIa and Class IIb devices (Class C and B IVDs): 3 years after the date of application;
- Class I devices (Class A IVDs): 5 years after the date of application;
- Reusable devices that shall bear the UDI Carrier on the device itself: 2 years after the date applicable for its respective class of devices.
Surveillance and Post Market
Clinical data, technical documentation and labeling must be updated. Furthermore, it will be necessary to review the processes including general safety and performance data, quality assurance, clinical trials, risk management, general compliance, traceability and post-market clinical follow-up that will require careful review, planning and updating for re-implementation in accordance with the new requirements.
With the new MDR, post-market surveillance is emphasized. This includes proactive performance monitoring of the recertification device, annual safety updates for high-risk class devices and prompt reporting of incidents.
Although the increase in the frequency of safety and performance reports may require significant additional resources for companies, these requirements can best identify potential problems at the beginning of the production cycle. Facing them can protect patients and reduce the producer's liability.
By the end of the transition process, any CE marked device must be fully compliant with the MDR. The UK Pharmaceutical Products Regulatory Agency (MHRA) has estimated that over half a million devices currently CE marked under Directives 93/42 / EEC and 90/385 / EEC will have to undergo the transition process and meet the new MDR.
Although many MDR requirements are similar to the current requirements in the field of MDD (Medical Device Directive) and AIMDD (Active Implantable Medical Devices Directive), the MDR is much more prescriptive and many devices will not have been deemed fully compliant with the new requirements.
As part of the MDR, the emphasis is on more in-depth reviews by notified bodies to confirm that manufacturers are fully compliant and that devices are fully supported by adequate data and technical documentation.